
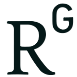

Hi 👋
I'm
Hemant Kumar Joon
About
From
Haryana, India
Lives In
Mannheim, Germany
Age
29
Website
github.com/hemantjoonSkills
Python
85%
R
80%
JAVA
50%
PHP
70%
HTML/CSS/JS
85%
Shiny
70%
Git & Github
80%
NGS
50%
Docker
70%
Machine Learning
60%
Nextflow
30%
SQL
85%
Android Development
70%
SAS
40%
Education
-
2011
10th
Air Force Bal Bharati School
New Delhi, India
Subjects: Science, Mathematics, Social Studies, English, Hindi, Sanskrit & IT
Achivements: Cleared with distinction
-
2013
12th
Air Force Bal Bharati School
New Delhi, India
Subjects: Biology, Mathematics, Physics, Chemistry & English
Achivements: Cleared with distinction
-
2016
BSc Zoology (Hons)
ARSD College, University of Delhi
New Delhi, India
Subjects: Zoology, Biochemistry, Bioinformatics & Biostatistics
Achivements: Cleared with distinction
Project: 2 weeks project to study Egret chronobiology
-
2018
MSc
Jamia Millia Islamia University
New Delhi, India
Subjects: Bioinformatics, Biostatistics, DBMS, & Programming and Problem Solving
Achivements: Entrance exam topper
Project: 4 month semester project in which we developed an android application to assist patient to predict the disease based on symptoms selected
-
2024
PhD Bioinformatics
International Centre for Genetic Engineering and Biotechnology
New Delhi, India
Topic: Integrated computational approaches to analyze and explore high-throughput cancer datasets
Research:
• Identify novel prognostic signatures for LUSC using publicly available microarray datasets.
• Develop a user-friendly tool for interactive exploration, interpretation and visualization of high throughput data.
Projects
VolFIS
Motivation: Probeset to multiple gene ID conversion and visualization of data were crucial steps for most bioinfomatics analysis
Aim: To develop a user friendly and highly customizable webserver catering ID conversion, file conversion and visualization of volcano and survival plots
Tech Stack: Frontend: HTML/CSS/JS; Backend: Python & R; Container: Docker; Server: Apache
Available: 14.139.62.220/volfis
AssayM
Motivation: To continuously monitor the appearances of mutations in the virus genome and mismatches in the PCR primers
Aim: To develop a web application to track the sensitivity of PCR primers on SARS-CoV-2 variants genome datasets
Tech Stack: Frontend: HTML/CSS/JS; Backend: PHP & Python; Database: MySQL; Container: Docker; Server: Apache
Available: assaymcovid19.kaust.edu.sa
iRSVPred
Motivation: To assist users to predict the variety of basmati rice seeds using an android phone and camera
Aim: Develop a user friendly android application and to provide a platform to perform basmati rice prediction using mobile itself
Tech Stack: IDE: Android Studio; Frontend: XML; Backend: JAVA, Python & PHP; Server: Apache
Available: play.google.com/store/apps/details?id=org.icgeb.irsvpred_2
Gene enrichment
Motivation: Gene enrichment is a crucial step in study and further analysis
Aim: To develop an automated pipeline from MCODE clustering to brite enrichment
Tech Stack: Python
Available: Available on request
SARS-CoV-2 | Shiny Dashboard
Motivation: Understanding shiny and build a dashboard
Aim: To develop a dashboard to visualize and filter SARS-CoV-2 data
Tech Stack: R
Available: hkjoon.shinyapps.io/sars-shiny-dashboard
Survival Analysis | Shiny Application
Motivation: Applying shiny for R for survival analysis
Aim: To develop a shiny application to perform survival analysis with various customization
Tech Stack: R
Available: hkjoon.shinyapps.io/survival-analysis-shiny-application
Codebase: github.com/hemantjoon/survival-analysis-shiny-application
Publications
-
VolFIS: A comprehensive web server for gene expression data ID conversion and data visualization
Hemant Kumar Joon, Anamika Thalor, and Dinesh Gupta
Numerous biological databases store data with varying gene identifiers, necessitating ID conversion in downstream genomic analysis. Standard downstream analysis in genomics involves differential gene expression and survival analysis, visualized through the volcano and survival plots. These plots are an easier way to explain the results of these analyses. So, it becomes necessary to create visually appealing, high-quality figures. While existing tools and R packages exist, they often require scripting knowledge, lack user-friendliness, and offer limited customization options to the users. Our study introduces VolFIS, a user-friendly web server addressing these challenges by providing user-friendly ID conversion and generating high-quality volcano and survival plots. It offers extensive customization options, enhancing downstream genomic analyses' interpretability. Further, it also allows direct file conversion for gene expression files, including options like NA value removal and merging identical gene names.
-
assayM COVID-19: a web application to track sensitivity of RT-PCR primers on COVID-19 variants
As of January 3, 2024, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has infected more than 773 million people and responsible for more than 6.99 million deaths globally (https://covid19.who.int/, accessed on January 3, 2023). At present, more than 16.40 million SARS-CoV-2 genome sequences are available on public databases such as Global Initiative of Sharing All Influenza Data (GISAID database), US National Center for Biotechnology Information (NCBI), etc., to analyze and detect the virus, understand the viral transmission and evolution mechanisms. However, rapidly evolving variants of the virus are increasingly becoming difficult to detect through the existing assays. Reverse Transcriptase – Polymerase Chain Reaction (RT-PCR) is the gold standard as diagnostic assays for the detection of COVID-19 and the specificity and sensitivity of these assays depend on the complementarity of the RT-PCR primers to the genome of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Virus mutates over time during replication cycles which will cause changes in the free energy upon primer/template binding, and will eventually affect the efficiencies of the detection assays. There is an urgent need to continuously monitor the appearances of mutations in the virus genome and mismatches in the PCR primers used in these assays. Here we present assayM COVID-19, a web application to track the sensitivity of PCR primers published by the World Health Organisation (WHO) or other custom-made primers on SARS-CoV-2 variants genome datasets along with the free energy variation as an indicator for the amplification efficiency. The web server can act as a single-stop platform for designing simple and LAMP-based primers, selection of the best WHO approved primers, primers binding site prediction, improving a primer’s binding efficiency, etc.
-
Machine learning analysis of lung squamous cell carcinoma gene expression datasets reveals novel prognostic signatures
Hemant Kumar Joon, Anamika Thalor, and Dinesh Gupta
LUSC is the second most prevalent subtype of lung cancer. LUSC patients are often diagnosed at an advanced stage and have poor prognoses. Thus, identifying novel biomarkers for the LUSC is of utmost importance. In this study, we downloaded multiple datasets from the NCBI GEO repository. Further, we merged all the datasets to construct a complete dataset comprising 963 samples and 20,031 genes. We also constructed a subset from this complete dataset having only known cancer driver genes (919 genes). Furthermore, we leverage RFE to obtain the top 10, 20, 30, 40, 50, and 60 features from both datasets. These top features were used as training features for the five ML classifiers: SVM, kNN, DT, RF, and XGBoost. For the complete and driver dataset, kNN performed comparatively better on the top 40 and top 50 gene features respectively. Out of these 90 gene features, 35 were found to be differentially regulated and the median risk score of these DEGs significantly stratified patients with better survival. Pathway enrichment analysis identified that these genes are associated with cell cycle, cell proliferation, and migration. We further validated our results using in-depth literature survey and found it to concord with the enrichment analysis.
-
Exploiting Machine Learning to Unravel Prognostic Biomarkers in Lung Cancer
Hemant Kumar Joon, Anamika Thalor, and Dinesh Gupta
Recent advances in machine learning have created opportunities to decode the genetic regulatory code and develop precision medicine for patients diagnosed with various kinds of diseases, including cancer. We hypothesize that machine learning and system biology could be applied to lung cancer gene expression profiles to identify novel biomarkers with therapeutic implications. Multiple gene expression profiles of lung cancer and normal lung tissue were obtained from the NCBI GEO datasets comprising>20k gene expressions. After pre-processing and merging the multiple datasets, the batch effect was corrected to purge the non-biological variations among the multiple datasets. Z-score standardization was employed on batch effect corrected dataset to get all the features on same scale. Further, a feature selection algorithm was employed to obtain the top 10, 20, 30, 40, 50, and 60 features from the complete gene expression profile having >20k genes to reduce the curse of dimensionality. Later, five machine learning algorithms were employed viz. support vector machine (SVM), k Nearest Neighbour (kNN), random forest (RF), decision tree (DS) and XGBoost on the above-selected features. kNN outperformed all the machine learning algorithms in training and on the external validation dataset downloaded from the same platform. The differential gene expression analysis (logFC > 1.5 and p-value < 0.05) was simultaneously employed on the complete dataset to identify the differentially expressed genes from the selected gene features obtained using machine learning. The next step is to identify prognostic biomarkers from the differentially expressed selected genes, using Kaplan Meier analysis
-
Machine learning assisted analysis of breast cancer gene expression profiles reveals novel potential prognostic biomarkers for triple-negative breast cancer
Anamika Thalor, Hemant Kumar Joon, Gagandeep Singh, Shikha Roy, and Dinesh Gupta
Tumor heterogeneity and the unclear metastasis mechanisms are the leading cause for the unavailability of effective targeted therapy for Triple-negative breast cancer (TNBC), a breast cancer (BrCa) subtype characterized by high mortality and high frequency of distant metastasis cases. The identification of prognostic biomarker can improve prognosis and personalized treatment regimes. Herein, we collected gene expression datasets representing TNBC and Non-TNBC BrCa. From the complete dataset, a subset reflecting solely known cancer driver genes was also constructed. Recursive Feature Elimination (RFE) was employed to identify top 20, 25, 30, 35, 40, 45, and 50 gene signatures that differentiate TNBC from the other BrCa subtypes. Five machine learning algorithms were employed on these selected features and on the basis of model performance evaluation, it was found that for the complete and driver dataset, XGBoost performs the best for a subset of 25 and 20 genes, respectively. Out of these 45 genes from the two datasets, 34 genes were found to be differentially regulated. The Kaplan-Meier (KM) analysis for Distant Metastasis Free Survival (DMFS) of these 34 differentially regulated genes revealed four genes, out of which two are novel that could be potential prognostic genes (POU2AF1 and S100B). Finally, interactome and pathway enrichment analyses were carried out to investigate the functional role of the identified potential prognostic genes in TNBC. These genes are associated with MAPK, PI3-AkT, Wnt, TGF-β, and other signal transduction pathways, pivotal in metastasis cascade. These gene signatures can provide novel molecular-level insights into metastasis.
-
Atomic Resolution Homology Models and Molecular Dynamics Simulations of Plasmodium falciparum Tubulins
Kanipakam Hema, Shahzaib Ahamad, Hemant Kumar Joon, Rajan Pandey, and Dinesh Gupta
Microtubules are tubulin polymers present in the eukaryotic cytoskeleton essential for structural stability and cell division that are also roadways for intracellular transport of vesicles and organelles. In the human malaria parasite Plasmodium falciparum, apart from providing structural stability and cell division, microtubules also facilitate important biological activities crucial for parasite survival in hosts, such as egression and motility. Hence, parasite structures and processes involving microtubules are among the most important drug targets for discovering much-needed novel Plasmodium inhibitors. The current study aims to construct reliable and high-quality 3D models of α-, β-, and γ-tubulins using various modeling techniques. We identified a common binding pocket specific to Plasmodium α-, β-, and γ-tubulins. Molecular dynamics simulations confirmed the stability of the Plasmodium tubulin 3D structures. The models generated in the present study may be used for protein–protein and protein–drug interaction investigations targeted toward designing malaria parasite tubulin-specific inhibitors.
Contact
Please, feel free to contact.
Location
Mannheim, Germany